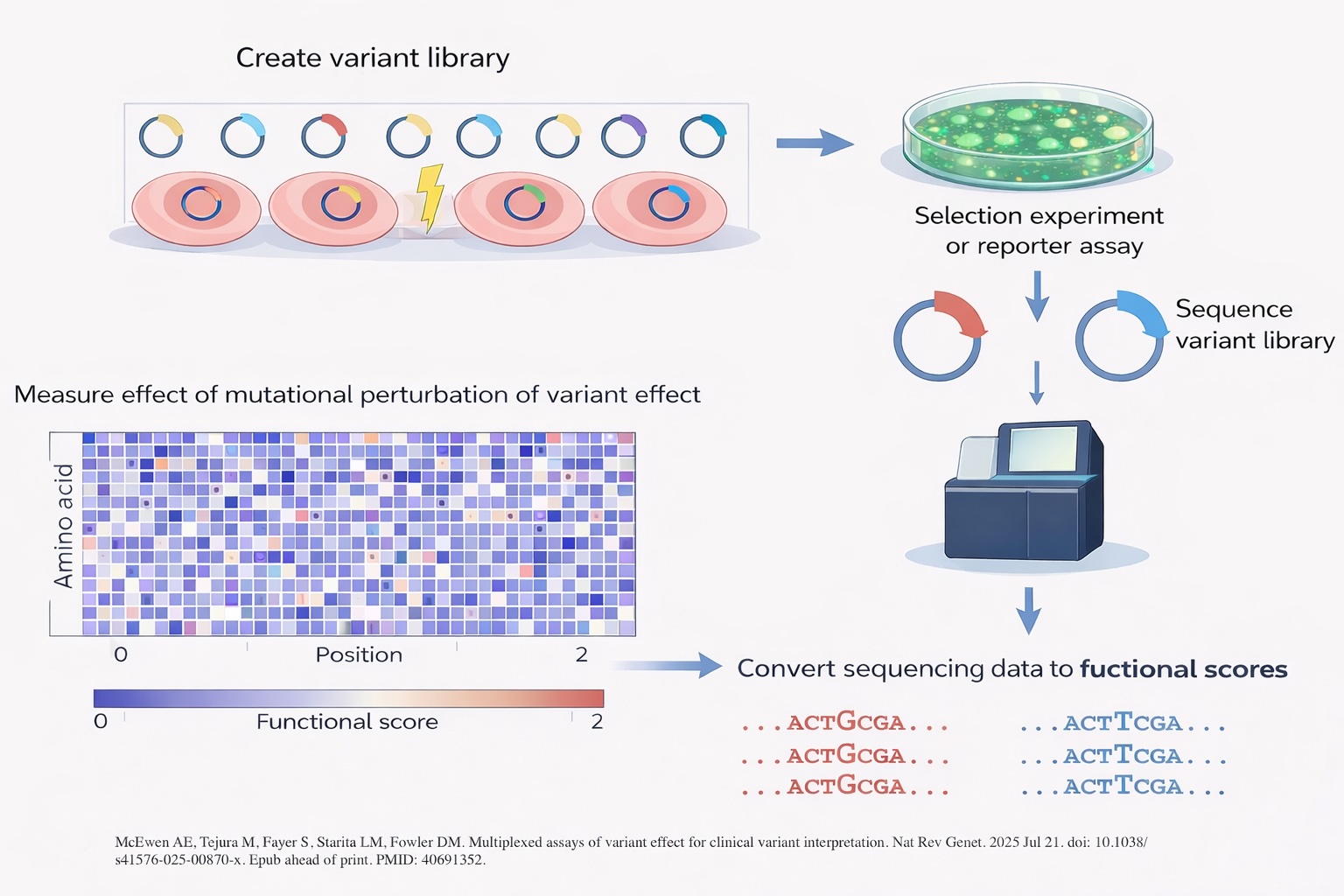
Multiplexed Assays of Variant Effect (MAVEs)
Despite widespread use of clinical sequencing—including targeted gene panels, whole-exome sequencing (WES), and whole-genome sequencing (WGS)—the diagnostic yield for inborn errors of immunity (IEIs) remains limited (~30%) ( Frontiers in Pediatrics, 2024 ). A major barrier is the interpretation of variants of uncertain significance (VUS). Our laboratory leverages multiplexed assays of variant effect (MAVEs) to systematically map genotype–function relationships and generate comprehensive functional atlases for key IEI genes.
Background
Although sequencing technologies have rapidly advanced, interpretation has become the primary bottleneck in genetic diagnosis. For IEIs in particular, many patients harbor rare or private variants whose functional consequences cannot be inferred from sequence data alone.
- The majority of variants submitted to clinical databases such as ClinVar are classified as variants of uncertain significance (VUS).
- The number of reported VUS continues to grow exponentially as sequencing expands (Nat Rev Genet, 2025).
- Testing variants individually is resource-intensive, time-consuming, and impractical at scale.
- Functional studies performed across different laboratories often yield inconsistent or non-comparable results.
Together, these challenges limit the clinical utility of sequencing and delay diagnosis for patients with suspected IEIs.
Genes and Pathways of Interest
- Our group is actively developing MAVEs for several high-yield IEI genes, selected based on clinical relevance, genetic constraint, and unmet diagnostic need.
- These genes span pathways involved in immune activation, regulation, and malignancy risk, and are frequently associated with high rates of VUS in clinical testing.
Our Approach
Our MAVE platform integrates:
- High-throughput variant generation, enabling saturation scale
- Physiologically relevant cellular systems tailored to gene-specific function
- Quantitative functional readouts that directly reflect immune phenotypes
- Rigorous statistical and computational analysis for reproducibility and clinical translation
Selected Publications
(As we are in the process of building our independent research program, representative publications from other groups illustrate conceptual and methodological approaches aligned with our work.)
Tabet DR, Coté AG, Lancaster MC, Weile J, Rayhan A, Fotiadou I, et al.
The functional landscape of coding variation in the familial hypercholesterolemia gene LDLR.
Science. 2025;eady7186.
DOI: 10.1126/science.ady7186
Walsh ZH, Frangieh CJ, Kothapalli N, Levy J, Heck CK, Melms JC, et al.
Scalable generation and functional classification of genetic variants in inborn errors of immunity to accelerate clinical diagnosis and treatment.
Cell. 2025;188(18):4861–4879.e27.
DOI: 10.1016/j.cell.2025.05.037